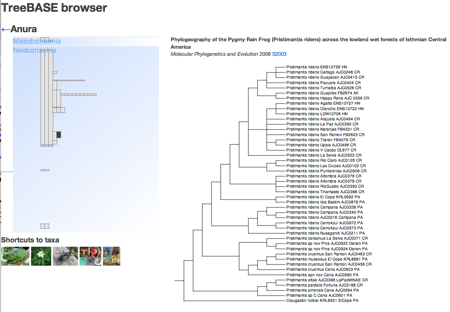
One thing which has always frustrated me about geophylogenies is how tedious they are to create. In theory, they should be pretty straightforward to generate. We take a tree, get point localities for each leaf in the tree, and generate the KML to display on Google Earth. The tedious part is getting the latitude and longitude data in the right format, and linking the leaves in the tree to the locality data.
To help reduce the tedium I've create a tool that tries to automate this as much as possible. The goal is to be able to paste in a NEXUS tree, and a table of localities, and get back a KML tree. Some publishers are making it easier to extract data from articles. For example, if you go to a paper such as
http://dx.doi.org/10.1016/j.ympev.2009.07.011 you will see a widget on the right labelled
Table download.

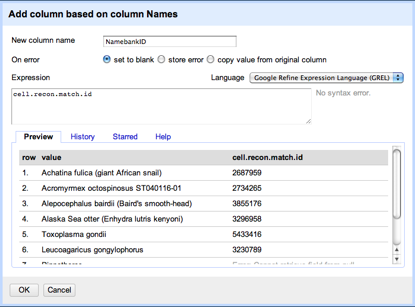
If you click on the
Find tables button you can download the tables in CSV format. In this case, Table 1 has latitude and longitude data for all the taxa in the tree in TreeBASE study
S10103. With some regular expressions we can figure out which column has the latitude and longitude data, and parse values like
(10°12′N, 84°09′W) to extract the numerical values for latitude and longitude.
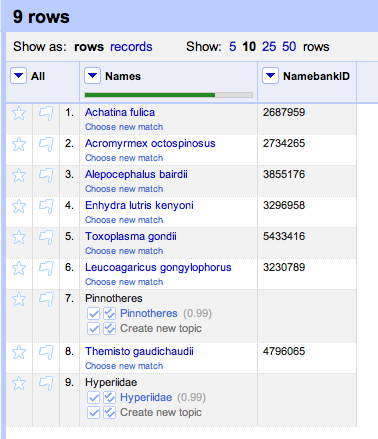
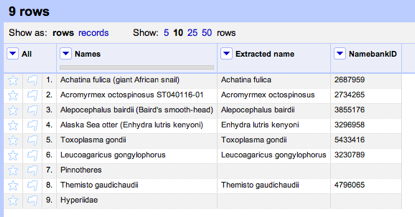
It is also pretty straightforward to be able to read a tree in NEXUS format and extract the taxon names. At this point we have two sets of names (those from the tree and those from the table) which might not be the same (in this case they aren't, we have "Craugastor cf. podiciferus FMNH 257672" and "FMNH 257672"). Matching these names up by hand would be tedious, but as described in
Matching names in phylogeny data files we can use maximum weighted bipartite matching to compute an optimal matching between the two sets of labels.
Create KML treeYou can try the Create KML tree tool at
http://iphylo.org/~rpage/phyloinformatics/kml/.
To get started, try it with the data below. In step 1 paste in the NEXUS tree, in step 2 paste in the table from the original paper. If all goes as it should, you will see a table displaying the matching, and the KML which you can save and open in Google Earth. If you have the
Google Earth Plug-in installed, then you should see the KML displayed on Google Earth in your web browser.
I've tested the tool on only a few examples, so there will be cases where it fails. It also assumes that every taxon in the tree has latitude and longitude values, and that the first column in the table is the taxon name (you'll need to edit the file if this is not the case).
Here is the tree used in the example...
#NEXUS
BEGIN TREES;
TRANSLATE
Tl254954 'Craugastor cf. podiciferus FMNH 257672',
Tl254956 'Craugastor cf. podiciferus FMNH 257653',
Tl254965 'Craugastor cf. podiciferus UCR 16356',
Tl254960 'Craugastor sp. A USNM 563039',
Tl254938 'Craugastor sp. A USNM 563040',
Tl254945 'Craugastor cf. podiciferus UCR 16360',
Tl254928 'Craugastor cf. podiciferus UCR 17439',
Tl254959 'Craugastor cf. podiciferus UCR 17462',
Tl254951 'Craugastor cf. podiciferus FMNH 257596',
Tl254967 'Craugastor sp. A FMNH 257689',
Tl254934 'Craugastor cf. podiciferus UCR 16355',
Tl254964 'Craugastor cf. podiciferus FMNH 257671',
Tl254963 'Craugastor cf. podiciferus UCR 16358',
Tl254952 'Craugastor cf. podiciferus UCR 18062',
Tl254926 'Craugastor cf. podiciferus UCR 17442',
Tl254968 'Craugastor sp. A FMNH 257562',
Tl254939 'Craugastor cf. podiciferus UCR 17441',
Tl254946 'Craugastor cf. podiciferus FMNH 257757',
Tl254942 'Craugastor cf. podiciferus MVZ 149813',
Tl254961 'Craugastor cf. podiciferus FMNH 257595',
Tl254969 'Craugastor cf. podiciferus UCR 17469',
Tl254932 'Craugastor cf. podiciferus MVZ 164825',
Tl254970 'Craugastor sp. A AJC 0891',
Tl254943 'Craugastor cf. podiciferus UCR 16357',
Tl254929 'Craugastor cf. podiciferus FMNH 257673',
Tl254950 'Craugastor cf. podiciferus FMNH 257756',
Tl254944 'Craugastor cf. podiciferus FMNH 257652',
Tl254953 'Craugastor cf. podiciferus UCR 16359',
Tl254931 'Craugastor cf. podiciferus UCR 17443',
Tl254940 'Craugastor stejnegerianus UCR 16332',
Tl254935 'Craugastor underwoodi UCR 16315',
Tl254958 'Craugastor cf. podiciferus UCR 16354',
Tl254966 'Craugastor sp. A AJC 0890',
Tl254949 'Craugastor cf. podiciferus FMNH 257758',
Tl254933 'Craugastor cf. podiciferus UCR 16361',
Tl254962 'Craugastor cf. podiciferus FMNH 257651',
Tl254948 'Craugastor cf. podiciferus FMNH 257670',
Tl254971 'Craugastor cf. podiciferus FMNH 257669',
Tl254936 'Craugastor cf. podiciferus FMNH 257550',
Tl254957 'Craugastor underwoodi USNM 561403',
Tl254947 'Craugastor cf. podiciferus FMNH 257755',
Tl254927 'Craugastor cf. podiciferus UCR 16353',
Tl254925 'Craugastor bransfordii MVUP 1875',
Tl254930 'Craugastor cf. podiciferus UTA A 52449',
Tl254955 'Craugastor tabasarae MVUP 1720',
Tl254941 'Craugastor cf. longirostris FMNH 257678',
Tl254937 'Craugastor cf. longirostris FMNH 257561' ;
TREE 'Fig. 2' = ((Tl254955,(Tl254941,Tl254937)),(((((Tl254954,Tl254942,Tl254933,Tl254948,Tl254971),((Tl254934,Tl254958,Tl254927),((Tl254964,Tl254929),Tl254930))),(((Tl254965,(Tl254963,Tl254943)),(Tl254959,Tl254969),(Tl254951,Tl254961)),((Tl254928,Tl254926,Tl254939,Tl254931),(Tl254952,Tl254932)))),((((Tl254956,Tl254936),Tl254946,Tl254950,(Tl254944,Tl254962),Tl254947),Tl254949),(Tl254945,Tl254953))),((((Tl254960,Tl254938),(Tl254970,Tl254966)),(Tl254967,Tl254968)),((Tl254940,Tl254925),(Tl254935,Tl254957)))));
END;
...and here is the table:
Taxon and institutional vouchera,Locality ID,Collection localityb,Geographic coordinates/approximate location,Elevation (m),GenBank accession number12S,16S,COI,c-myc
1. UTA A-52449,1,"Puntarenas, CR","(10°18′N, 84°48′W)",1520,EF562312,EF562365,None,EF562417
2. MVZ 149813,2,"Puntarenas, CR","(10°18′N, 84°42′W)",1500,EF562319,EF562373,EF562386,EF562430
3. FMNH 257669,1,"Puntarenas, CR","(10°18′N, 84°47′W)",1500,EF562320,EF562372,EF562380,EF562432
4. FMNH 257670,1,"Puntarenas, CR","(10°18′N, 84°47′W)",1500,EF562317,EF562336,EF562376,EF562421
5. FMNH 257671,1,"Puntarenas, CR","(10°18′N, 84°47′W)",1500,EF562314,EF562374,EF562409,None
6. FMNH 257672,1,"Puntarenas, CR","(10°18′N, 84°47′W)",1500,EF562318,None,EF562382,None
7. FMNH 257673,1,"Puntarenas, CR","(10°18′N, 84°47′W)",1500,EF562311,EF562343,EF562392,None
8. UCR 16361,3,"Alejuela, CR","(10°13′ N, 84°22′W)",1930,EF562321,EF562371,EF562375,EF562431
9. UCR 16353,4,"Heredia, CR","(10°12′N, 84°09′W)",1500,EF562313,EF562349,None,EF562420
10. UCR 16354,4,"Heredia, CR","(10°12′N, 84°09′W)",1500,EF562315,EF562363,None,EF562418
11. UCR 16355,4,"Heredia, CR","(10°12′N, 84°09′W)",1500,EF562316,EF562366,None,EF562419
12. UCR 18062,6,"Heredia, CR","(10°10′N, 84°06′W)",1900,EF562302,EF562342,EF562395,None
13. UCR 17439,5,"Heredia, CR","(10°09′N, 84°09′W)",2000,EF562298,EF562341,EF562387,EF562427
14. UCR 17441,5,"Heredia, CR","(10°09′N, 84°09′W)",2000,EF562299,EF562345,EF562388,EF562429
15. UCR 17442,5,"Heredia, CR","(10°09′N, 84°09′W)",2000,EF562300,EF562337,EF562385,EF562422
16. UCR 17443,5,"Heredia, CR","(10°09′N, 84°09′W)",2000,EF562301,EF562340,EF562384,EF562428
17. UCR 17462,5,"Heredia, CR","(10°09′N, 84°09′W)",2000,EF562309,EF562355,EF562406,EF562440
18. UCR 17469,5,"Heredia, CR","(10°09′N, 84°09′W)",2000,EF562310,EF562334,EF562405,EF562414
19. MVZ 164825,7,"Heredia, CR","(10° 05′N, 84° 04′W)",2100,EF562303,EF562346,EF562381,EF562423
20. UCR 16357,8,"San José, CR","(10°02′N, 83°57′W)",1600,EF562306,EF562339,EF562400,EF562433
21. UCR 16358,8,"San José, CR","(10°02′N, 83°57′W)",1600,EF562307,EF562370,EF562412,EF562415
22. UCR 16356,8,"San José, CR","(10°01′N, 83°56′W)",1940,EF562308,EF562329,None,None
23. UCR 16359,10,"San José, CR","(9°26′N, 83°41′W)",1313,EF562297,EF562369,EF562396,None
24. UCR 16360,10,"San José, CR","(9°26′N, 83°41′W)",1313,EF562296,EF562368,None,EF562434
25. FMNH 257595,9,"Cartago, CR","(9°44′N, 83°46′W)",1600,EF562304,EF562338,EF562408,None
26. FMNH 257596,9,"Cartago, CR","(9°44′N, 83°46′W)",1600,EF562305,EF562335,None,EF562416
27. FMNH 257550,11,"Puntarenas, CR","(8°47′N, 82°59′W)",1350,EF562294,EF562330,EF562393,EF562443
28. FMNH 257651,11,"Puntarenas, CR","(8°47′N, 82°59′W)",1350,EF562291,EF562367,EF562402,EF562435
29. FMNH 257652,11,"Puntarenas, CR","(8°47′N, 82°59′W)",1350,EF562288,EF562364,EF562390,None
30. FMNH 257653,11,"Puntarenas, CR","(8°47′N, 82°59′W)",1350,EF562292,EF562354,EF562392,EF562438
31. FMNH 257755,11,"Puntarenas, CR","(8°46′N, 82°59′W)",1410,EF562289,EF562344,EF562379,None
32. FMNH 257756,11,"Puntarenas, CR","(8°46′N, 82°59′W)",1410,EF562290,EF562347,EF562377,EF562413
33. FMNH 257757,11,"Puntarenas, CR","(8°46′N, 82°59′W)",1410,EF562293,EF562352,EF562383,EF562437
34. FMNH 257758,11,"Puntarenas, CR","(8°46′N, 82°59′W)",1410,EF562295,EF562348,EF562397,EF562436
35. USNM 563039,12,"Chiriquí, PA","(8°48′N, 82°24′W)",1663,EF562284,EF562356,EF562389,EF562445
36. USNM 563040,12,"Chiriquí, PA","(8°48′N, 82°24′W)",1663,EF562285,EF562350,EF562391,EF562439
37. AJC 0890,12,"Chiriquí, PA","(8°48′N, 82°24′W)",1663,EF562282,EF562351,EF562398,EF562444
38. MVUP 1880,12,"Chiriquí, PA","(8°48′N, 82°24′W)",1663,EF562283,EF562358,EF562399,EF562442
39. FMNH 257689,12,"Chiriquí, PA","(8°45′N, 82°13′W)",1100,EF562287,EF562353,EF562407,EF562446
40. FMNH 257562,12,"Chiriquí, PA","(8°45′N, 82°13′W)",1100,EF562286,EF562357,EF562410,EF562441
41. USNM 561403,N/A,"Heredia, CR","(10°24′N, 84°03′W)",800,EF562323,EF562361,EF562378,None
42. UCR 16315,N/A,"Alejuela, CR","(10°13′N, 84°35′W)",960,EF562322,EF562362,EF562394,None
43. UCR 16332,N/A,"San José, CR","(9°18′N, 83°46′W)",900,EF562325,EF562360,EF562411,AY211320
44. MVUP 1875 fitzingeri group,N/A,"BDT, PA","(9°24′N, 82°17′W)",50,EF562324,EF562359,None,AY211304
45. MVUP 1720,N/A,"Coclé, PA","(8°40′N, 80°35′W)",800,EF562326,EF562332,EF562401,EF562424
46. FMNH 257561,N/A,"Chiriquí, PA","(8°45′N, 82°13′W)",1100,EF562327,EF562331,None,EF562426
47. FMNH 257678,N/A,"Chiriquí, PA","(8°45′N, 82°13′W)",1100,EF562328,EF562333,EF562404,EF562425

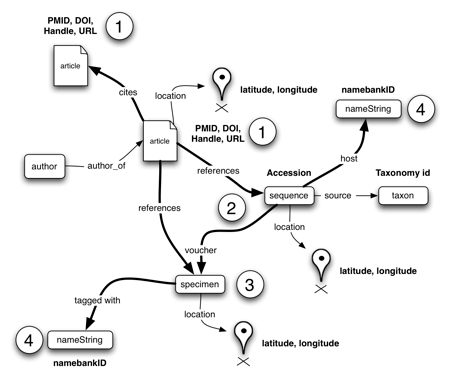
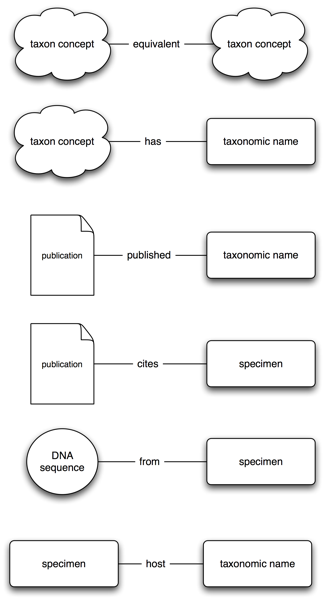
 Brief update on
Brief update on 
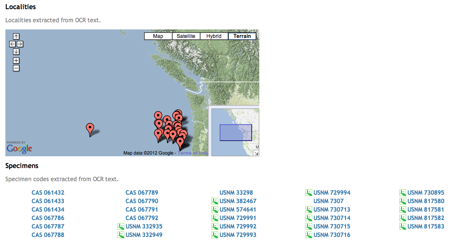
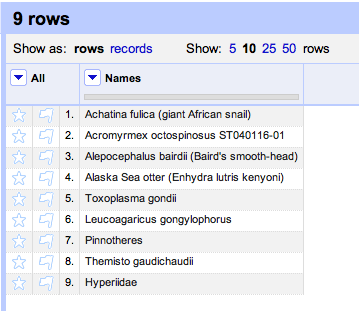
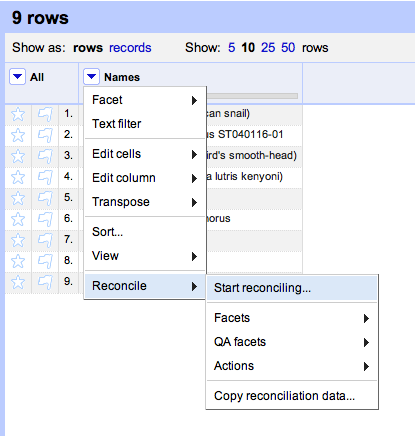
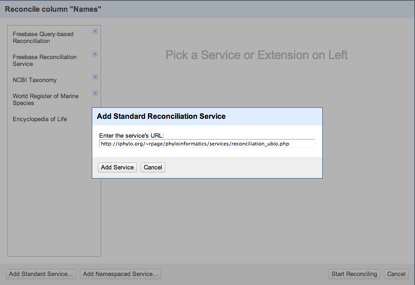
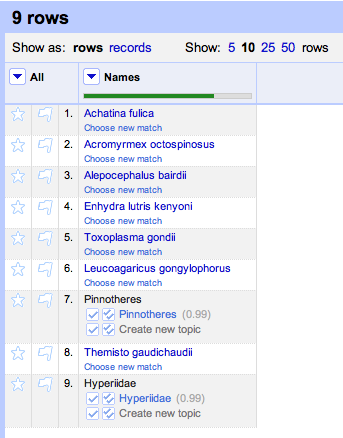
 Duplicate records are the bane of any project that aggregates data from multiple sources.
Duplicate records are the bane of any project that aggregates data from multiple sources. 



 The Encyclopedia of Life have announced the
The Encyclopedia of Life have announced the